
James Townsend
Root Canal: Reassessment of the Animal Tree of Life and Its Implications
ISSUE 59 | TAXONOMY | DEC 2015


Among a handful of researchers studying the early history of animal life a scientific debate is playing out, rather quietly, with stakes ranging from the contents of high school biology textbooks to our very concept of how evolution proceeds on earth. At issue is one of the great questions of evolutionary biology: in what order did the “basal phyla,” or the extant groups of animals thought to be among the oldest on earth, branch from each other as animals diversified on earth? The question boils down to which group of animals branched off from the rest first and is thus the “sister group” to all animal groups that were derived later. Recent DNA sequence data, resulting from the recent revolution in sequencing cost, suggest that Ctenophora, or “comb jellies,” a phylum of gelatinous plankton, may have a claim to being that group. This modern result, coming from our ability to both sequence DNA and model its evolution in supercomputers, contrasts sharply with over a century of traditional biological thought (some might say dogma) that Porifera (sea sponges) were the oldest-branching group on earth. In the process of evaluating these claims, scientists have been forced to grapple with various potential biases underlying the sciences of taxonomy (the classification of animals), phylogeny (discerning evolutionary relationships between these categories), and the perception of what is even possible during evolution of life on earth.
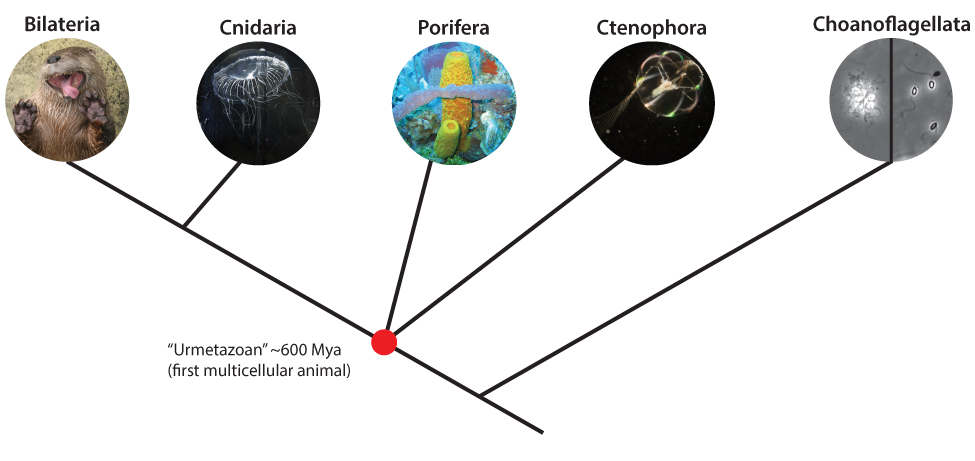
Figure 1: Schematic of early animal relationships. Modern debate centers on the phylogenetic placement of Ctenophora vs. Porifera as the first surviving group of animals to differentiate from the rest. The identity of this sister group to all other animals has profound implications for the nature of the story of animal evolution.
Sponges are bottom-dwelling filter-feeding creatures with a dizzying variety of irregular, geometric forms, many of which might seem more like a marine plant or mold to a casual observer, but that nonetheless are animals. Ctenophores are perhaps less familiar. These planktonic1 “comb jellies” consist of only about 150 known species living suspended throughout the world’s oceans. They are principally composed of a gelatinous material that is surrounded by a diaphanously thin layer of cells, leading many to see them as just another kind of “jellyfish.” However, what most people imagine when they hear “jellyfish” is in fact some member of another more familiar phylum, Cnidaria. Cnidarians include everything from Portuguese man-o-war jellyfish to the corals composing coral reefs, and are defined by the presence of stinging cells on their tentacles that they use to stun and capture their prey. In contrast, ctenophores use sticky cells called colloblasts to ensnare their food. And whereas cnidarian jellyfish move by pulsing their bell-shaped bodies, ctenophores use their eight characteristic rows of ciliated “combs” to paddle their way through the water. Ctenophores are therefore easily distinguishable as a distinct group from cnidarians by their comb rows, lack of sting, and various other, smaller distinctive physiological features.
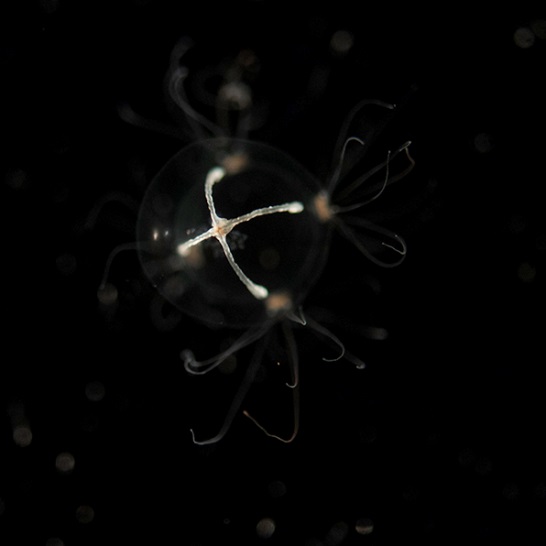

Figure 2: Morphological similarity of ctenophores and cnidarians. A cnidarian (unidentified hydrozoan, at left) and a ctenophore (Pleurobrachia piletus, right). These animals are both ~1 cm in diameter, and were caught in the same location in southern New Jersey. The ctenophore is easily distinguished by the radially arranged “comb rows” running the length of its body, but the overall similarity is apparent.
Sponges, cnidarians, and ctenophores together constitute the major members of the basal phyla2, inferred to be among the oldest animal groups with representatives still living on earth. All of these animals are predominantly marine organisms, and have held a certain scientific appeal and interest since marine biological sampling began in earnest in the 18th century. In spite of this, like many marine animals, they remain understudied relative to, say, rats or humans. There are still many open questions in their basic physiology.
I study the biochemistry and structure of the amorphous, gelatinous portion of ctenophores, called the mesoglea, which occupies 10,000 times more space than do the cells of the animal, which is the part we might more readily point to as “alive.” There have been a few isolated attempts to investigate this aspect of ctenophore anatomy but nothing conclusive, and nothing published on the matter in decades. It’s not clear exactly what ctenophore mesoglea is made of or how it is arranged by or produced in the animal, though the means exist today to investigate both matters quite thoroughly. As a young Ph.D. student, this seemed like an interesting and tractable subject for a thesis. But this project only became more interesting as the old debate about the precise layout of the basal animal lineage—who came first and using what logic—flared up again in the light of new genomic data on ctenophores. The result was that my once-understudied phylum of interest is suddenly a hot topic for many. The hunger for more detailed information about these animals’ obscure physiology gave my project and others’ a new rationale. Uncovering data that help to dislodge the current stalemate in the discussion of basal phyla lineage and move the debate forward in any direction would be a service to the field and a personal accomplishment for the researchers involved. It’s simply unclear under the circumstances what data will be enough to manage this.
In order to appreciate the origins and implications of this situation, a discussion of the history and practice of taxonomy is in order.
The main task of biology could be said to be one of rendering comprehensible a living world of interminable complexity. The scientific process too is messy and often fractally difficult and nuanced. Every level of scrutiny recapitulates the difficulties of the ones above it. The goals of science—accurate, succinct models to explain this complexity—is all too often just out of reach. There is usually one more complication, one more special case. Sometimes special cases become major complications or, if you’re lucky, important discoveries in their own right. Usually, they just remind scientists of the scale of their work and keep grad students busy on weekends.
Taxonomy is no different. The process of giving a name to each new species and hierarchically nested groups of species requires a researcher to decide where the boundaries between species are, which then entails the need to decide how to define boundaries, or whether they even exist.
Linnaeus, the founder of modern taxonomic practice and creator of the now-ubiquitous binomial classification system (Genus species naming), believed in the 18th century that the task of cataloging all the species in the world could be completed in a generation or two, and that it was his god-given mission to make it happen. He believed there were on the order of tens of thousands of species. He rarely left Europe. As Linnaeus’ students and subsequent generations of scientists explored the world’s forests, plains, and oceans—not to mention the microscopic universes inside every drop of pond water—the number of species ballooned, and then exploded. The estimated total number of species on Earth quickly became a nebulously high number—millions, tens of millions—of which only a fraction are known to science, and a still smaller fraction have been studied in any great depth.
But if a list of species, whether it has ten thousand entries or ten million, is to be of much use to anyone, it must be organized in a way that makes it navigable, or at least provides some means to naïvely browse it. An English dictionary is indexed by the letters of the alphabet. Ideally, species would be organized into similarly well-defined categories representative of some biological reality. Since the dawning of the epoch of evolution in biology the organizing principle of species organization has been taken to mean that species should be organized by their phylogenetic relationships—their groupings and positions relative to one another on the evolutionary “tree of life.” Prior to the mid-20th century, taxonomists were forced to rely upon body shape and shared traits during embryonic development as rough proxies for evolutionary relatedness. Similar physical traits, such as number of limbs and where they were connected to the body, even in different species, suggested a kind of relatedness that was at least useful for building a phylogenic tree, even as the species’ precise evolutionary history was pure speculation. These methods work surprisingly well for inferring evolutionarily relatedness of groups that split apart from one another fairly recently, such as mites that live associated with humans and their livestock.
Arguably the greatest shortcoming of this method was its overreliance on scientists’ preconceived notions and (lack of) ability to predict of how the process of evolution could and would play itself out to currently observed endpoints. Browsing very early phylogenetic trees is a good illustration of this. Errors, egregious to even a young biology student, abound. Typically these take the form of categorization errors based on flawed heuristics, such as judging sessile animals to be plantlike in origin, or on creeping anthropocentrism, wherein animals similar to humans in some way are thought to be “above” (or later branching than) others that are less similar. What was always needed was some measurable character common to all living things, preferably one that could be analyzed by a consistent toolkit of techniques. Quite fortunately for the field, just such a character exists. Its discovery and elucidation in the 20th century is without a doubt one of the greatest scientific advances in history. I’m referring, of course, to the genome and the substrate on which it’s encoded, DNA.3
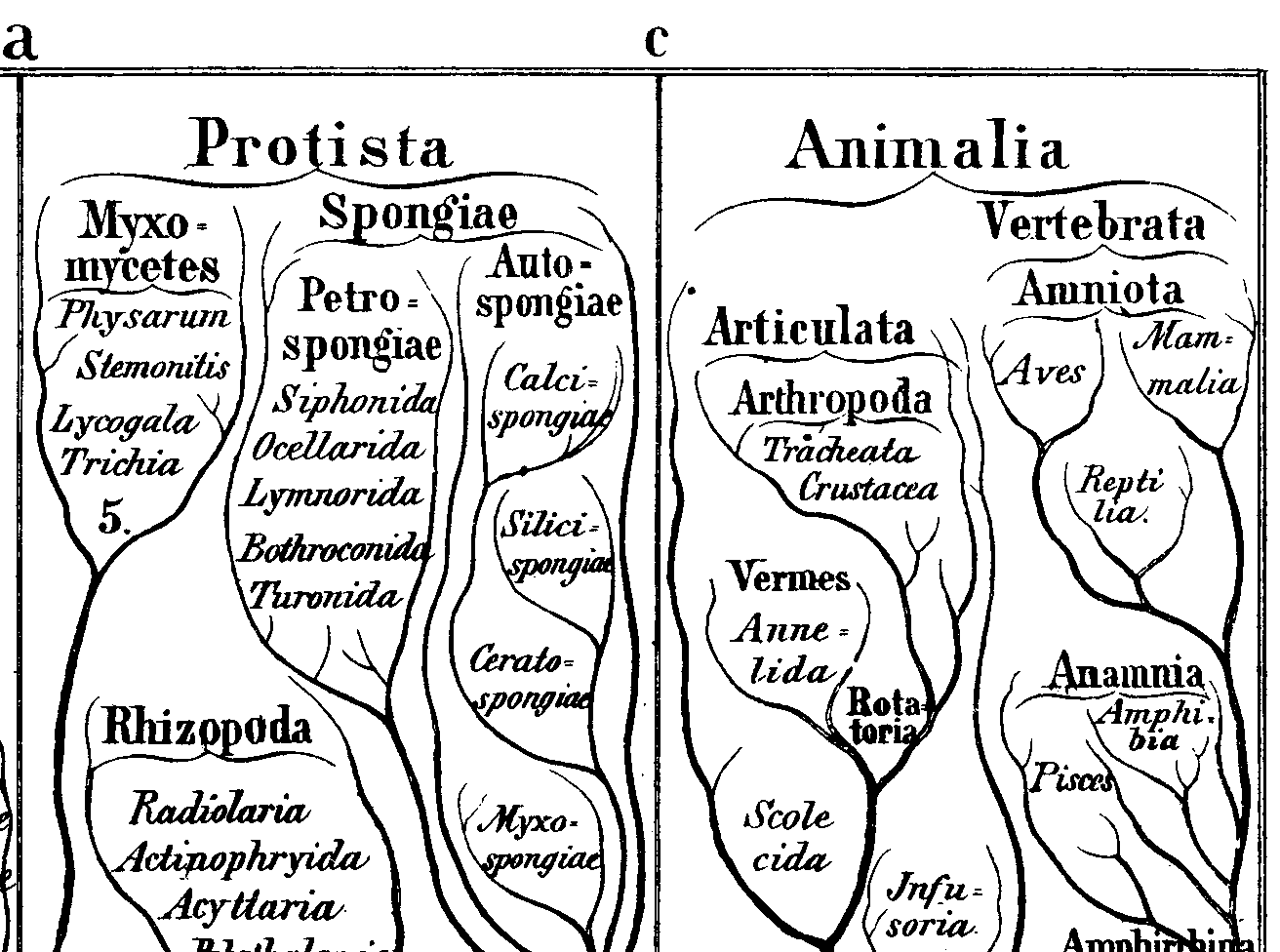
Figure 3: Closeup of an early phylogenetic tree from Earnst Haekel's Generelle Morphologie der Organismen (1866) in which sponges (Spongiae) are not identified as animals. Interestingly, in this engraving, the phylogenetic tree is literally rendered as a tree.
If two DNA sequences are similar, they are assumed to be related to one another, and the degree of similarity assumed to be a measure of their relatedness. The genomes (the complete DNA sequences) of a parent and daughter organism, for instance, are typically somewhere between identical and highly similar overall. And while there are genetic variations among a population’s various individuals, they are still typically readily distinguishable from individuals of a different species. Genetic variations can accumulate through random fluctuations in a population’s DNA sequences; when individuals with some of these random fluctuations are more likely to produce offspring for the next generation than others, the makeup of a population’s DNA shifts – this process writ large is evolution.
To understand evolution at long timescales, biologists have collected reference sequences from different organisms’ genomes; the goal is to determine the relatedness between these genomes by comparing their sequences and scoring them by similarity. Species are then grouped according to these scores. These groups of species can be related to other groups of species in a similar fashion, and so on.
A longstanding convention in biology is to organize species’ groupings into dendrograms or phylogenetic trees that recall the early “tree of life” diagrams of Darwin and Haekel 4. Any two species or groups of species on the tree are connected by a node that represents their last common ancestor. The depths of nodes within a tree can be used to estimate the time since the groups of species diverged from their common ancestor. Species or groups that branch off relatively earlier in a tree are referred to as basal, while those that appear relatively later are derived. Terms such as “higher” or “lower,” while in common use even among some scientists, are not accurate. A tree is not a hierarchy or a ladder. All extant species have been evolving for the same amount of time, and the tree is agnostic to anything other than their relationships to one another in time.
Many readers will be familiar with the idea of taxonomic ranking (kingdom, phylum, class, etc.). This system, popularized by Linnaeus, seeks to group species based on their observable characteristics into increasingly fine-grained categories. Perhaps some even had to memorize some version of this ordering system in a biology class, a la “The mitochondrion is the powerhouse of the cell…” That the system is subject to rote memorization would imply that it is a closed issue in science, but the truth is a bit messier, in part thanks to the advent of modern DNA sequencing and molecular phylogeny.
While it is the case that there is a standardized taxonomic nomenclature in science complete with professional groups that supervise it, it is important to remember that this nomenclature is essentially arbitrary and exists for the convenience of humans and doesn’t necessarily represent a more fundamental truth about life on earth. A “family” of sculpins or a “class” of cnidarians based on apparent physiology is just one potential organizational scheme of many. Shifting to organizing species by genetic sequence similarity necessitates a reexamination of the logic behind constructing these subgroups.
When scientists moved to categorizing organisms by the similarity of their DNA, the number and depth of nodes in these new “molecular” trees became a product of a series of mathematical operations done on the data. These new trees may or may not recapitulate the taxonomic rankings based on physiology, which arises from genes, but indirectly so, so correspondence between the two isn’t a given. Likewise, a molecular phylogenetic tree need not retain the depth of the conventional taxonomic ordering system. That is, there may be more categories or fewer. A species may be monotypic—the only member of a taxonomic group—many layers deep in classification space. 5 It’s possible for the old conventions of hierarchy to become somewhat meaningless under the right circumstances.
Thinking about this serves to underscore an important point. All science is model-making, and all models contain assumptions. Making fewer assumptions is a great goal. In shifting to using sequence data to construct phylogenies, we drop certain assumptions about phylogeny and are potentially closer to an ideal taxonomic organization system for it: we acknowledge that distinct genomic inputs can lead to similar phenotypic outputs and that subtle genetic differences can lead to surprising physiological differences. By looking at the molecular medium of heredity, we get that much closer to the sort of biological reality that would make for a good indexing system.
Unfortunately, that newly-recognized subtlety and complexity makes the task of evaluating the reasonability of biological models that much more complicated and subtle.
This brings us to the present scientific debate on the phylogenetic order of the early animals. The evolution of animals has held a special place in professional and public discussion of evolutionary biology for some time, likely due to some combination of familiarity, immediacy, and narcissism. As with cosmologists and the Big Bang, or anthropologists and the earliest civilizations, “The Beginning” of animal life holds an irresistible appeal. Scientists understand well enough that before the rise of multicellular animals or Metazoa, there existed some ancestral, single-celled eukaryote (the domain of life comprising all organisms not in achaebacteria or eubacteria). A group of single-celled marine eukaryotes called choanoflagellates likely represents the protists present today that are most similar to these ancestral eukaryotes. This makes choanoflagellates the nearest “outgroup” to Metazoa—an important consideration for generating phylogenetic trees from genomes and a major point of contention in this debate. Of great interest to biologists is what traits were instrumental in transitioning from single-celled organisms, perhaps even from colonies of single-celled organisms (as is seen in some species of choanoflagellates), to highly integrated multicellular animals. Theories abound, from enhanced signaling and coordination between cells to the elaboration of existing systems of cellular polarity. But all of this theorizing, until very recently, has been predicated on the idea that the earliest group of animals to branch off and become distinct was sea sponges. The implications of this assumption are worth a bit of scrutiny.
First, the positioning of Porifera as the most basal group of animals is not without historic merit. The logic behind this positioning is relatively extensive: sponges lack many of the complex traits and physiological features associated with more recently derived groups of animals, such as nerves, muscles, and body symmetry. Sponges possess feeding cells called choanocytes, which resemble single-celled choanoflagellates. Genetic studies and, eventually, the first published sponge genome, further supported the notion that the morphologically indistinct sponges had branched off prior to the development of animals with more symmetric bodies. The idea of “sponges first” has been relatively uncontested since the early 20th century and dozens of prominent researchers base some part their current research programs on this notion.
However, one problem with these analyses was that they were necessarily done in the absence of much substantive data on ctenophores. Even though Ctenophora has been recognized as a distinct phylogenetic group for centuries, and long presumed to be among the older groups of animals, ctenophores spent much of the 19th and 20th centuries admired for their aesthetic value and scrutinized for their complex behaviors and feeding ecologies, but not considered to be particularly key to understanding the origins of Metazoans. The reasons for this are many. First, there are simply very few species of ctenophores around: about 150 species of ctenophores are known to modern science, many described only relatively recently and in exotic locations such as the deep ocean or in Arctic waters. Compare this to sponges, of which there are thousands of species, or to cnidarians, a group numbering in the tens of thousands of species. While several species of ctenophores are quite prolific and inhabit wide swaths of the world’s oceans, it’s easy enough to view them as more of an interesting curiosity than a potential missing link. Second, most ctenophores are incredibly fragile. A bit of a lame excuse by some measures, but I can attest that they’re often not too easy to catch and maintain by today’s standards, making them that much more difficult for scientists of the past to study.
Finally, the relative “complexity” of ctenophores caused scientists to assume that the group had branched off more recently. Ctenophores have nervous systems, muscles, body symmetry, as well as complex behaviors traditionally associated with more recently derived animals. What’s more, they are morphologically similar to the famed cnidarian “jellyfish” (mostly from the class Scyphozoa). Therefore, ctenophores and cnidarians were until recently commonly considered as part of a single taxonomic group called coelenterates. These were thought to be separate from and older than bilaterians (the large, but apparently correct group containing all animals with bilateral symmetry), but more recently derived than sponges. This persisted for some time, until around 2008, when the first major ctenophore sequencing studies started to appear.
The appearance of data on ctenophore genetics, first as snippets of DNA sequence, and then in the form of two complete genomes of different ctenophore species, was the key to kicking off a new phase in the phylogenetic study of the basal phyla. Given a rich body of genetic information on Bilateria, Cnidaria, Porifera, and Ctenophora, the discussion of their phylogenetic ordering could proceed in the light of a more complete body of input knowledge.
The researchers who had collected the genomic data on the ctenophore Mnemiopsis leidyi had already begun to use this data for enhanced phylogenetic tree-building in 2008. Their initial analysis turned up a very surprising result: the data strongly suggested that ctenophores, in spite of their “complex” features, were actually the sister group to all other animals— ctenophores seemed to be the basal metazoans that branched off first. This was in complete contrast not only to the body of evidence suggesting that sponges held this position, but also in apparent violation of a critical idea in evolutionary biology, the concept of parsimony. The principle of parsimony directs scientists not to invent complex explanations where a simpler one will do.
For if ctenophores are the sister group to all other animals, and sponges were derived later on, it implies one of two things: that the common ancestor of ctenophores and other animals with nerves and muscles and symmetry (Cnidaria and Bilateria) also possessed these features, and that they were simply lost in the sponge lineage, or that these “complex” physiological features somehow evolved twice, independently of each other. The first explanation is problematic because it further implies that the first multicellular animal, the “Urmetazoan” from which all other animals were derived, was actually much more sophisticated in physiology than previously thought possible. The second explanation is perhaps an even more difficult pill to swallow: for nerves and muscles and symmetry to all to have evolved multiple times within Metazoa would suggest they are much more readily obtained than previously assumed.
The result ignited major debates in fields as diverse as neuroscience and developmental biology. An enormous historical precedent hangs in the balance, and with it, reputations.

Figure 4: Mnemiopsis leidyi, the first ctenophore to have its genome sequenced.

Figure 5: Amphimedon queenslandica, the first sponge to have its genome sequenced. The morphological simplicity of this animal has served as the anchor point for quite a bit of research. (adapted from Adamska et al. 2007)
Subsequent years of research have brought yet more sequence data on ctenophores that have tended to further support the initial hypothesis of ctenophores as sister group to all other animals, as well as extensive re-analysis of the data that already exist, with varying results. Depending on the particular mathematical models used to estimate the evolutionary relationships contained in the genetic sequence data, different investigators have recovered different phylogenetic relationships. Sometimes sponges are basal; other times, ctenophores. A major focus of the public debate is on what models for analyzing the genetic sequences and constructing trees are actually valid given the circumstances, and which are more likely to introduce statistical artifacts. For example, some researchers have criticized the organisms used for comparison to animals used in the initial genome studies; by including species too distant from Metazoa it is possible to make all early-branching things look more related than they actually are.
For other researchers (myself included), the question has shifted to trying to make sense of the surprising, but possible, model of ctenophores being the basal animals. This result does at first seem to be unparsimonious. However, it is also possible that scientists stand before a gaping flaw in our basic assumptions about how evolution works. As it turns out, there are plenty of gaps in the knowledge of ctenophore and sponge physiology and development where the means to address this complex issue might live. Renewed interest in filling in these gaps by labs the world over has already begun yielding some interesting results. As an example, take the unusual workings of the ctenophore nervous system, which has only one neurotransmitter in common with our own nerves (glutamate) and employs a variety of still-mysterious neuropeptides. Similarly, the comparison between the jelly of ctenophores and the jelly of cnidarian jellyfish is proving to be superficial; at a chemical level, it turns out that this is more like comparing the flippers of penguins and the fins of fish than it is to comparing one simple jelly to another.
The ctenophores-as-basal-animals position requires two shifts in scientific thinking. First, it requires scientists to understand and trust algorithms that model the evolution of genetic sequence over more traditional morphological methods of understanding evolutionary relatedness. This shift in thinking has been in the works for several decades now, but this instance indicates a significant surge in acceptance of genomics as the way forward for evolutionary biology. This is likely to be part changing of the guard, as young scientists employ newer, more computationally intensive modes of analysis, and part paradigm shift, as proponents of molecular evolution solidify their position within the broader study of biology.
The second way in which the basal positioning of ctenophores signals a sea change in evolutionary thinking is that it has the real potential to completely upset the anthropocentric apple cart that has been hard for even the most devoted evolutionists to completely part with. That is to say, prior to this surprising result, evolution was typically conceptualized as a process of building up complexity. Animal life was thought to become more and more sophisticated over time, with new features being added at every branching point. This scheme eventually leads, predictably, to humans and our ilk, presumably among the more complex species on the block. But to acknowledge the validity of the basal placement of ctenophores is to acknowledge that “complexity” as we understand it is a potentially flighty thing, hard to pin down. Given this result, we are highly aware that evolution is a process that only optimizes locally across time, and under which it is quite easy to lose complex features so long as they are not adaptive.
Alternately, we may be forced to acknowledge that traits we had been reading as complex, owing to their relationship to humans (e.g. having a nervous system), are not actually quite as unique or formidable an evolutionary triumph as we once thought, or even at all. This too has been in the works for some time now. Increasing levels of taxonomic sampling and close examination of the natural world have revealed that it is often all too easy to lose “complex” functionality and have that loss become fixed within a species, even if it seems strictly bad (e.g. humans’ inability to produce vitamin C endogenously). These insights, themselves often driven by molecular analysis, have likewise made scientists more accepting of the basic concepts needed to accommodate the present debate.
Whether or not the basal positioning of ctenophores will stand in the long run or not is anyone’s guess at this point. The debate among key researchers is far from over. But the symbolic impact of the debate and underlying hypothesis is clear: biologists are now more willing to break ranks with the phylogenetic models and methods of the past than ever before. This is a good thing if it does indeed help to remove the potential for certain unconscious biases, which it probably will. But the genomic age of biology has undoubtedly already brought with it a new litany of models to be evaluated for validity, utility, and yes, bias.
A potential existential question for the field of molecular evolution arising from the basal metazoan debate is “Is this problem even soluble?” Is there a hard limit to what scientists can deduce of evolutionary history from the information present on earth today, and are they close to that limit? This is something of concern to all scientists, but the potential for anxiety is increased here because what’s being studied is more like a cold case murder mystery than fixing a broken TV, which is to say, it’s a bit harder to know when you’ve figured it out. What constitutes confirmation of solving the mystery of what extant animal group is the oldest? The general standard could be said to be when the obtained results jive with all other accepted data on the topic, and when the results are useful for modeling or understanding future results. So, when molecular phylogenetic clustering in basal animals recovers accepted details further out along the tree, and when the relationships in these basal animal trees are helpful for making sense of the phenotypic differences between different sponges, ctenophores, cnidarians, and so on—perhaps then we can say we have a working model.
The kicker is that basal animal genomes and basal animal physiology are still surprisingly underexplored. There’s an incredible laundry list of topics that have never been touched or have been investigated a bit but never published. Evolutionary biology may be on the cusp of a Kuhnian paradigm shift (or not), but there are not enough data to really assess the matter. Until there are, this internecine debate is bound to continue and quite possibly grow in scope as more scientists join in. For what it’s worth, this is far from the first time anyone has worried that a branch of science had reached a fundamental limit or end, though it has never yet proven to be the case. Hopefully, sufficient scrutiny will bring some resolution, possibly even enlightenment.
Acknowledgements: I’d like to thank Dr. Alison Sweeney for her input in editing this piece.
Further reading:
Original ctenophore genome publications:
Ryan et al., 2013:
https://www.sciencemag.org/content/342/6164/1242592.abstract
Moroz et al., 2014: http://www.nature.com/nature/journal/v510/n7503/full/nature13400.html
Sponge genome publication:
Srivastava et al., 2010:
http://www.nature.com/nature/journal/v466/n7307/full/nature09201.html
Examples of articles reexamining extant basal phyla genome data:
Whelan et al., 2015:
http://www.pnas.org/content/112/18/5773.full.pdf
Pisani et al. 2015:
http://www.pnas.org/content/early/2015/11/24/1518127112.full.pdf
1 An organism is planktonic if it lives in the water column and drifts with the current. Plankton can be microscopic or enormous. Ctenophores tend to be on the scale of centimeters in length.
2 For simplicity, I am not discussing Placozoa here, though they are fascinating and ultra-strange to the point of being a potential edge case.
3 And yes, RNA.
4 The only illustration in Darwin’s On the Origin of Species is a lithograph of a simple model tree used to demonstrate the idea variation arising out of common descent. Haekel took the concept and ran with it, coining the term for the science of generating trees, “phylogeny,” that is still used today.
5 For example, the entire animal phylum Placozoa has only one member, Trichoplax adherens.

The Hypocrite Reader is free, but we publish some of the most fascinating writing on the internet. Our editors are volunteers and, until recently, so were our writers. During the 2020 coronavirus pandemic, we decided we needed to find a way to pay contributors for their work.
Help us pay writers (and our server bills) so we can keep this stuff coming. At that link, you can become a recurring backer on Patreon, where we offer thrilling rewards to our supporters. If you can't swing a monthly donation, you can also make a 1-time donation through our Ko-fi; even a few dollars helps!
The Hypocrite Reader operates without any kind of institutional support, and for the foreseeable future we plan to keep it that way. Your contributions are the only way we are able to keep doing what we do!
And if you'd like to read more of our useful, unexpected content, you can join our mailing list so that you'll hear from us when we publish.